
Din cuprinsul articolului
Aproximativ 5-10% dintre cancere au o componentă ereditară puternică, iar identificarea mutațiilor genetice poate transforma radical abordarea medicală, de la prevenție la tratament. În România, testele genetice devin tot mai accesibile, iar inițiative precum programele Primăriei București oferă screening gratuit pentru mii de persoane.
În timp ce majoritatea cancerelor apar din întâmplare, consecință a acumulării de mutații de-a lungul vieții, există familii unde boala lovește repetat, generație după generație. Pentru aceste persoane, cancerul nu e doar ghinionul unor celule deviate, ci o predispoziție scrisă în ADN, moștenită de la părinți. Vestea bună: știința a ajuns să identifice genele responsabile și să ofere teste care pot anticipa riscul cu decenii înainte ca boala să apară.
Datele recente arată că între 5 și 10% din totalul cancerelor au o cauză ereditară clară, determinate de mutații transmise din generație în generație. În România, accesul la testarea genetică se îmbunătățește progresiv. Primăria Municipiului București a lansat în 2024 un program pilot care oferă teste genetice gratuite pentru 1.500 de beneficiari, acoperind costuri de până la 4.000 de lei per persoană, după ce au primit indicație de la medicul oncolog.
Laboratoarele private precum Synevo, MedLife, Regina Maria și SYNLAB au introdus paneluri extinse de testare genetică care analizează simultan zeci de gene asociate cu riscul de cancer. Prețurile variază de la câteva sute de lei pentru testele de bază până la peste 3.000-4.000 de lei pentru panelurile comprehensive care evaluează 20-60 de gene.
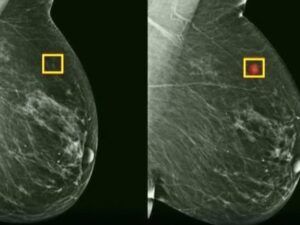
Cancerul de sân și ovar: genele BRCA în prim-plan
Cel mai cunoscut exemplu de cancer ereditar rămâne cel de sân și ovar, asociat cu mutațiile genelor BRCA1 și BRCA2. Aceste gene supresoare tumorale au rol esențial în repararea ADN-ului deteriorat. Când funcționează corect, ele opresc celulele cu defecte să se multiplice necontrolat.
O femeie care moștenește o mutație BRCA1 are între 55% și 72% șanse să dezvolte cancer mamar de-a lungul vieții, comparativ cu aproximativ 12% în populația generală. Pentru BRCA2, riscul variază între 45% și 69%. La cancerul ovarian, cifrele sunt și mai îngrijorătoare: 39-44% pentru BRCA1 și 11-17% pentru BRCA2, față de sub 1% în medie.

Celebrele cazuri ale actriței Angelina Jolie și ale lui Christina Applegate au adus în atenția publică decizia drastică a mastectomiei profilactice – îndepărtarea preventivă a sânilor pentru a preîntâmpina boala. Jolie a ales această intervenție după ce testarea genetică a relevat că are o mutație BRCA1, estimându-i-se un risc de 87% pentru cancer mamar.
Dar BRCA1 și BRCA2 nu sunt singurele gene implicate. Cercetările au identificat alte gene precum PALB2, CHEK2, ATM, BRIP1, RAD51C și RAD51D, responsabile de aproximativ 10% din cancerele ovariene ereditare. Testele moderne de tip multigenic analizează simultan toate aceste variante.
Important de reținut: mutațiile BRCA cresc riscul și pentru alte tipuri de cancer. Bărbații purtători ai unei mutații BRCA2 au un risc de 20 de ori mai mare pentru cancer de prostată și de 10 ori mai mare pentru cancer pancreatic. Ambele gene sunt asociate și cu cancere ale stomacului, laringelui și chiar cu melanom.
Sindromul Lynch: inamicul tăcut al colonului
Cunoscut și sub denumirea de cancer colorectal non-polipozic ereditar (HNPCC), sindromul Lynch e cel mai frecvent tip de cancer de colon transmis genetic, răspunzător pentru 1-3% din totalul cancerelor colorectale. Spre deosebire de alte forme de cancer colonic, acesta nu produce sute de polipi intestinali, ceea ce îl face mai greu de detectat prin metodele clasice.
Sindromul Lynch apare din cauza mutațiilor în genele de reparare a ADN-ului: MLH1, MSH2, MSH6, PMS2 și EPCAM. Aceste gene funcționează ca un sistem de corectare a erorilor care apar în mod natural când celulele se divid. Când sistemul e defect, erorile se acumulează și celulele devin canceroase.
Persoanele cu sindrom Lynch au un risc cumulat de până la 80% de a dezvolta cancer colorectal până la 70 de ani. Femeile au și un risc de 60% pentru cancer endometrial. Boala lovește devreme – vârsta medie de diagnostic e de 44 de ani, cu un deceniu mai timpuriu decât în cazurile sporadice.
Lista cancerelor asociate e extinsă: stomac, intestin subțire, pancreas, vezică urinară, căi biliare, piele și chiar tumori cerebrale (în sindromul Turcot, o variantă rară). Mutațiile în gena MSH2 par să fie mai frecvent legate de aceste manifestări extracolonice.
Testarea pentru sindromul Lynch se recomandă persoanelor care au cel puțin trei rude cu cancer colorectal, dintre care una e rudă de gradul întâi cu celelalte două, afectând minimum două generații, iar cel puțin o persoană a fost diagnosticată înainte de 50 de ani. Acești parametri se numesc „criteriile Amsterdam”.
Persoanele diagnosticate cu sindrom Lynch trebuie să înceapă colonoscopiile de screening la 20-25 de ani și să le repete la fiecare 1-3 ani. Femeile necesită și monitorizare ginecologică prin ecografie transvaginală și biopsie endometrială începând de la 25-35 de ani.
Polipoza adenomatoasă familială: când colonul se acoperă de polipi
Polipoza adenomatoasă familială (PAF) e dramatică prin manifestările ei: sute sau chiar mii de polipi adenomatoși apar în colon și rect, de obicei în adolescență. Fără tratament, aproape 100% dintre pacienți dezvoltă cancer colorectal până la 35-40 de ani.
Boala se datorează mutațiilor genei APC, situată pe cromozomul 5. Aproximativ 70-75% din cazuri sunt moștenite de la unul dintre părinți, iar în 25-30% mutația apare spontan în cursul dezvoltării embrionare. Gena APC produce o proteină care reglează creșterea celulară; când lipsește sau e defectă, celulele se multiplică haotic.
Există și o formă atenuată a bolii, cu mai puțini polipi (în medie 30) și debut mai târziu. Indiferent de formă, colectomia totală – îndepărtarea chirurgicală a colonului – devine inevitabilă pentru prevenirea cancerului. Intervenția se planifică de obicei între 17 și 20 de ani, înainte ca polipii să malignizeze.
PAF se poate asocia cu sindromul Gardner, caracterizat prin tumori osoase benigne (osteoame), tumori ale glandelor sebacee și tumori desmoide – formațiuni agresive ale țesuturilor moi care pot apărea după operații. O altă variantă, sindromul Turcot, asociază polipoza cu tumori cerebrale.
Screening-ul genetic pentru APC se recomandă în familiile afectate încă de la 10-11 ani, urmat de rectosigmoidoscopie regulată. Depistarea precoce permite planificarea intervențiilor chirurgicale înainte de apariția cancerului.
Unii pacienți cu polipoza adenomatoasă prezintă mutații în gena MUTYH (transmisă autozomal recesiv – ambii părinți trebuie să fie purtători), în genele POLE și POLD1 (responsabile de polimeraza ADN), sau în gena NTHL1. Aceste forme mai rare necesită abordări personalizate.
Melanomul familial: când pielea poartă risc genetic
Aproximativ 5-10% dintre melanoame apar într-un context familial, iar identificarea genelor responsabile a evoluat substanțial în ultimii 20 de ani. În 1994, cercetătorii au descoperit că mutațiile genei CDKN2A (care codifică proteina p16) sunt principala cauză a melanomului ereditar, prezente în aproximativ 40% din familiile cu risc crescut.
CDKN2A funcționează ca un frână a ciclului celular, oprind celulele să se dividă când nu trebuie. Mutațiile acestei gene permit multiplicarea necontrolată și apar în special la persoane cu debut precoce al melanomului (sub 45 de ani), cu melanome multiple sau cu istoric familial semnificativ.
Purtătorii unei mutații CDKN2A au un risc de 28-67% de a dezvolta melanom până la 80 de ani, diagnosticul apărând la o vârstă medie de 33-45 de ani, comparativ cu 53-61 în populația generală. Dar surpriza vine din faptul că acești pacienți au și un risc de 28% pentru cancer pancreatic, față de sub 1% în populația generală.
Gena CDK4, identificată în 1996, e implicată în aproximativ 17 familii la nivel global. Mutațiile ei (în special la codonul 24) produc un fenotip similar cu CDKN2A: debut precoce, melanome multiple și naevi atipici. Recent, s-au descoperit și alte gene mai rare: MITF, BAP1, TERT, POT1, ACD și TERF2IP, fiecare contribuind la sub 1% din cazurile familiale.
Testarea genetică pentru melanom se recomandă persoanelor cu peste 50 de naevi, cu trei sau mai multe melanome, sau cu istoric familial de melanom plus cancer pancreatic sau astrocitom. Screening-ul dermatologic trebuie să înceapă devreme și să fie regulat, iar protecția solară devine obligatorie.
Sindromul Li-Fraumeni: când riscul e multiplu
Sindromul Li-Fraumeni e poate cel mai dramatice dintre cancerele ereditare: e asociat cu un spectru larg de tumori maligne și un risc cumulat de cancer care atinge aproape 100% la femei și 75% la bărbați până la 70 de ani. Jumătate din aceste cancere apar înainte de 40 de ani.
Cauza e mutația genei TP53, considerată „gardianul genomului”. Proteina p53 produsă de această genă monitorizează integritatea ADN-ului și declanșează fie repararea deteriorărilor, fie moartea programată a celulelor iremediabil afectate. Când p53 nu funcționează, celulele cu defecte continuă să se multiplice.
Cincisprezece tipuri de cancer se asociază cu sindromul Li-Fraumeni, dar cinci domină tabloul clinic: sarcoamele de țesuturi moi (20% din cazuri), osteosarcoamele (15%), tumorile cerebrale (13%), cancerul de sân cu debut timpuriu (25%) și carcinomul adrenocortical – un cancer rar al glandelor suprarenale.
Distribuția pe vârste e revelatoare: tumorile cerebrale, sarcoamele și carcinomul adrenocortical apar în prima decadă de viață, osteosarcoamele și leucemiile în adolescență, cancerul de sân între 20-40 de ani, iar cancerele de colon, pancreas și prostată după 50 de ani.
Diagnosticul sindromului Li-Fraumeni se stabilește când o persoană are un sarcom înainte de 45 de ani, o rudă de gradul întâi cu cancer înainte de 45 de ani, și o altă rudă de gradul întâi sau doi cu cancer timpuriu sau sarcom la orice vârstă. Testarea genetică confirmă prezența mutației TP53.
Managementul pacienților cu sindrom Li-Fraumeni e complex și necesită monitorizare extensivă: RMN cerebral și RMN corp întreg anual, mamografie sau RMN mamar la fiecare șase luni (de la 20-25 ani la femei), ecografie abdominală regulată, colonoscopie la fiecare 2-3 ani și examinări dermatologice complete. Protocolul Toronto, dezvoltat specific pentru această afecțiune, recomandă examinări fizice complete la fiecare 6-12 luni încă din copilărie.
Alte sindroame ereditare mai rare
Lista cancerelor ereditare nu se oprește aici. Sindromul von Hippel-Lindau, cauzat de mutații ale genei VHL, predispune la tumori renale, feocromocitom și hemangioblaostoame cerebeloase. Neoplazia endocrină multiplă (MEN) produce tumori ale glandelor endocrine, în special tiroida, paratiroida și glandele suprarenale.
Sindromul Cowden, determinat de mutații PTEN, crește riscul pentru cancer tiroidian, de sân și endometrial. Retinoblastomul ereditar, cauzat de mutații RB1, produce tumori oculare la copii. Sindromul cancerului gastric difuz ereditar, asociat cu mutații CDH1, duce la cancer gastric agresiv cu debut foarte timpuriu.
Când trebuie să te gândești la testarea genetică
Medicii recomandă consiliere genetică și, eventual, testare în următoarele situații:
- Diagnostic de cancer la vârstă tânără (sub 45-50 de ani, în funcție de tipul de cancer)
- Cancere multiple la aceeași persoană
- Cancer la ambii sâni sau la ambii ovare
- Istoric familial cu mai multe cazuri de același tip de cancer
- Cancer rar (adrenocortical, sarcoame, cancer de sân masculin)
- Apartenență la grupuri etnice cu risc crescut (de exemplu, evreii Ashkenazi pentru BRCA)
- Un membru al familiei cu mutație genetică cunoscută
Testarea genetică se face printr-o simplă recoltare de sânge, urmată de analiza ADN-ului prin secvențiere de nouă generație (NGS) și tehnici complementare precum MLPA pentru detectarea deletiilor și duplicațiilor mari. Rezultatele vin de obicei în 2-4 săptămâni.
Ce înseamnă un rezultat pozitiv
Identificarea unei mutații genetice nu înseamnă automat că vei dezvolta cancer, ci doar că ai un risc semnificativ mai mare decât populația generală. Deciziile ulterioare depind de multe variabile: tipul specific de mutație, vârsta, sexul, istoricul personal și familial.
Opțiunile includ:
Supraveghere intensificată: screening-uri mai frecvente și mai complete, începute la vârste mai tinere. De exemplu, femeile cu mutații BRCA încep mamografiile și RMN-urile mamare de la 25-30 de ani, nu de la 40 de ani ca în populația generală.
Chirurgie profilactică: mastectomia și ooforectomia (îndepărtarea ovarelor) reduc dramatic riscul de cancer la purtătoarele de mutații BRCA. Studiile arată că mastectomia bilaterală reduce riscul cu peste 90%, iar ooforectomia cu 80-90% pentru cancer ovarian.
Medicamente preventive: tamoxifenul și raloxifenul pot reduce riscul de cancer mamar la femei selectate. Aspirina e studiată pentru prevenția cancerului colorectal în sindromul Lynch.
Planificare familială: persoanele cu mutații ereditare au 50% șanse să le transmită copiilor. Diagnosticul genetic preimplantațional permite selectarea embrionilor fără mutație în cadrul fertilizării in vitro.
Viitorul testării genetice în cancer
Cercetarea avansează rapid. Studiile actuale explorează noi gene de susceptibilitate, încearcă să înțeleagă de ce unii purtători de mutații dezvoltă cancer și alții nu, și identifică factori modificatori genetici și de mediu care influențează riscul.
Tehnologiile de secvențiere a genomului întreg devin tot mai accesibile, permițând identificarea unor mutații rare sau în gene încă necaracterizate complet. Inteligența artificială începe să fie folosită pentru a prezice riscul individual pe baza combinației de variante genetice, nu doar a unei singure mutații.
Testarea genetică germinală (a ADN-ului moștenit) începe să fie complementată de testarea tumorală somatică. Uneori, analiza țesutului tumoral relevă mutații care sugerează o predispoziție ereditară, chiar dacă pacientul nu avea un istoric familial evident.
Aspecte etice și psihologice
Decizia de a face testare genetică nu e simplă. Unii oameni preferă să nu știe, temându-se de anxietatea pe care o poate genera un rezultat pozitiv sau de potențiala discriminare (deși legislația europeană protejează împotriva acesteia în asigurări și angajare).
Testarea ridică și întrebări despre obligația morală de a informa rudele. Dacă descoperi că ai o mutație BRCA, frații, părinții și copiii tăi au 50% șanse să o aibă și ei. Ești obligat să le spui? Ce faci dacă nu vor să știe?
Pentru copii, testarea genetică e recomandată doar pentru sindroame care necesită intervenții în copilărie (precum Li-Fraumeni sau polipoza adenomatoasă familială). Pentru sindroame cu manifestare la adult (ca BRCA), se preferă amânarea testării până când copilul poate lua singur decizia informată.
De reținut
Testarea genetică pentru cancerele ereditare nu e pentru toată lumea, dar pentru familiile afectate poate fi transformatoare. Identificarea unei mutații deschide uși către prevenție, screening precoce și, când boala apare totuși, către tratamente personalizate mai eficiente.
În România, accesul la aceste teste se îmbunătățește treptat. Laboratoarele private oferă paneluri comprehensive, iar inițiativele de sănătate publică încep să recunoască valoarea testării genetice. Provocarea rămâne educația – atât a medicilor, pentru a identifica pacienții candidați, cât și a publicului, pentru a înțelege când testarea e relevantă și ce înseamnă rezultatele.
Pentru cei cu istoric familial semnificativ de cancer, discuția cu un medic genetician sau un consilier genetic e primul pas. Întrebările „De ce noi?” și „De ce se repetă în familia noastră?” pot avea acum răspunsuri concrete – și, mai important, soluții care salvează vieți.
Notă: Acest articol are scop informativ și educativ. Deciziile privind testarea genetică trebuie luate în urma discuțiilor cu medicul curant și, preferabil, cu un specialist în genetică medicală sau un consilier genetic. Testarea genetică fără consiliere adecvată poate avea consecințe psihologice negative și poate duce la interpretări eronate.
Surse:
ARPIM – Testarea genetică în cancerele ereditare (Primăria București – 4.000 lei/beneficiar)
NCBI – Lynch Syndrome GeneReviews
JAMA – Cancer Risks Associated With Germline Mutations in MLH1, MSH2, and MSH6
PubMed – Low prevalence of germline CDKN2A and CDK4 mutations in patients with early-onset melanoma
Wiley Online Library – Genetics of familial melanoma: 20 years after CDKN2A
NCBI – Li-Fraumeni Syndrome GeneReviews
NCBI StatPearls – Li-Fraumeni Syndrome
Cleveland Clinic – Li-Fraumeni Syndrome: Symptoms, Causes & Outlook
Nature – Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes
MedlinePlus Genetics – Li-Fraumeni syndrome
PMC – Inherited TP53 Mutations and the Li-Fraumeni Syndrome
National Comprehensive Cancer Network (NCCN) GuidelinesJAX – Lynch Syndrome Factsheet




